5. Aqueous phase chemistry
Because the heterogeneous processes described in Chapter 4 are so fast, we can consider aqueous chemistry to be equilibrium chemistry for periods longer than a few seconds. Recall that the development and dissipation of clouds is typically tens of minutes, and sometimes more. So equilibrium aqueous chemistry should be very applicable to issues such as acid rain.
5.1 Aqueous-phase
equilibria
In the aqueous phase, several chemical species rapidly dissociate into ions. The time constant achieving equilibrium between the adsorbed gas and its ions in solution is typically less than 1 ms. Thus, we can assume equilibrium for our calculations.
5.1.1 Water.
Water ionizes in to the hydrogen ion, H+, and the hydroxide ion, OH-. In reality, H+ quickly attaches to another water to form the hydronium ion, H3O+.
At equilibrium, H2O Û H+ + OH- (or H2O + H2O Û H3O+ + OH-1)
![]()
Kw’ = 1.82 x 10-16 M at 298 K. But [H2O] = 55.5 M = 55.5 mole/L, which is much larger than the concentration of the ions and is essentially constant. We multiply by [H2O] to get the ion product of water: Kw’[H2O]
Kw = 1.0 x 10‑14 M2 at 298 K. In pure water, [H+] = [OH‑].
The pH of water is defined as pH = - log10[H+], and is 7.0 at 298 K. When pH is less than 7, the solution is acidic. When pH is greater than 7, the solution is basic.
5.1.2. Carbon
Dioxide / Water Equilibrium.
CO2 hydrolyzes in water to form CO2·H2O, which is sometimes called CO2(aq). The following equilibria are established:
CO2(g) + H2O Û CO2·H2O
CO2·H2O Û H+ + HCO3- (bicarbonate ion) Kc1
HCO3- Û H+ + CO32- (carbonate ion) Kc2
Now that we have the equations, we can set up the equilibria. and then solve the simultaneous equations:
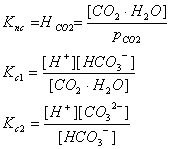
We want to get all of these in terms of things that we can actually measure.
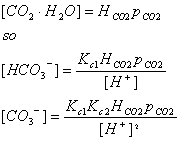
We are interested in several quantities, including the total dissolved carbon:
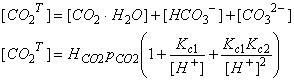
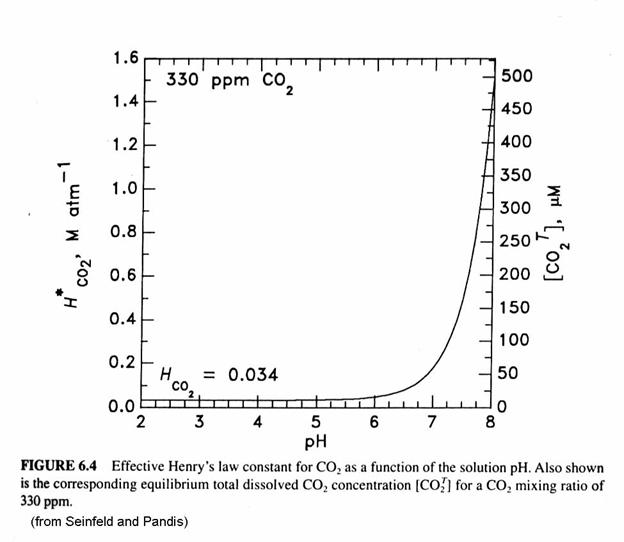
More carbon is dissolved than we would expect from Henry’s Law alone. We can determine an effective Henry’s Law constant, which will be greater than the actual Henry Law Constant:

Note that the effective Henry’s Law Constant is greater than the Henry’s Law Constant. This difference results from the carbon that is dissolved in forms other than [CO2∙H2O], which is in equilibrium with the gas-phase CO2. We see that the effective Henry’s Law constant is dependent on pH through the hydronium ion concentration, [H+] = [H3O+], as can be seen in following figure. The total amount of dissolved carbon is a strong function of pH, or [H+]. The effective Henry’s Law Constant for a pH of 8 is more than 10,000 times larger than HCO2.
In order to quantitative numbers, we must determine the ion concentrations. We can do this by assuming that the solution started out neutral and that the total positive ions exactly balance the total negative ions:
[H+] = [OH-] + [HCO3-] + 2[CO32-]
Using the equilibrium relations derived before, we can rewrite this expression in terms of equilibrium coefficients, the Henry’s Law Constant, and the partial pressure of CO2:
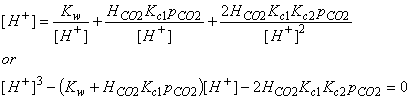
For a temperature of 298 K and a CO2 mixing ratio of 350 ppmv, the pH of a solution of water and dissolved CO2 will be 5.6. Because atmospheric CO2 is always there, a pH of 5.6 is considered the pH of pure rainwater.
5.1.3. Sulfur dioxide / water equilibrium
After SO2 is absorbed into cloud drops (H(298 K) = 1.2 M atm-1), it ionizes (step 3) to form bisulfite (HSO3-) and sulfite (SO32-) ions by the reactions:
(1) SO2(g) + H2O <==> SO2.H2O
(2) SO2.H2O <==> H+ + HSO3- ; HSO3- is the bisulfite ion
(3) HSO3- <==> H+ + SO32- ; SO32- is the sulfite ion
(4) H2O <==> H+ +
These reactions have the equilibrium constants:
HSO2= [SO2.H2O ]/pSO2
Ks1 = [H+][HSO3-]/[SO2.H2O]
Ks2 = [H+][SO32- ]/[HSO3-]
Kw = [H+][OH-] ; This is called the ion product of water: Kw = 10-14 M2 @ 298 K.
The concentrations of the dissolved species are:
[SO2.H2O] = HSO2 pSO2
[HSO3-] = Ks1[SO2.H2O]/[H+] = HSO2 Ks1 pSO2/[H+]
[SO32- ] = Ks2[HSO3-]/[H+] = HSO2Ks1Ks2 pSO2 / [H+]2
Let total sulfur that is in the IV oxidation state in the aqueous solution be written as:
[S(IV)] = [SO2.H2O] + [HSO3-] + [SO32- ]
Writing all of the concentrations as a combination of constants and [H+]:
[S(IV)] = HSO2 pSO2 {1 + Ks1/[H+] + Ks1 Ks2 /[H+]2}
Therefore, we can write total dissolved sulfur as:
[S(IV)] = HS(IV)* pSO2
The effective Henry's Law coefficient is:
HS(IV)* = HSO2 {1 + Ks1/[H+] + Ks1 Ks2 /[H+]2}
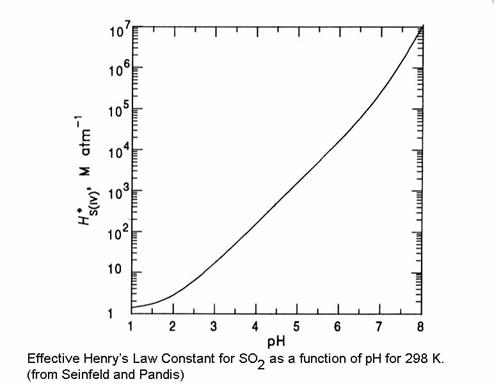
Note that H*S(IV) is always greater than HS(IV). Note also that the total dissolved sulfur is dependent on the pH.

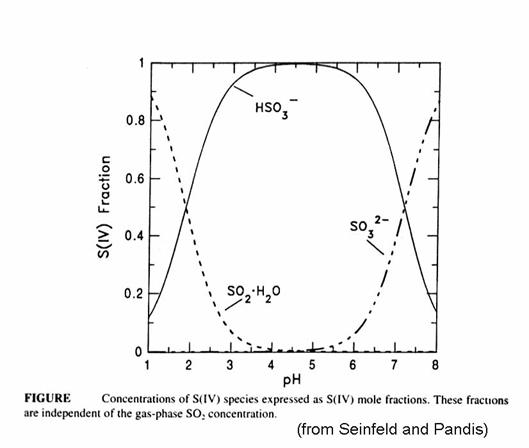
The fraction of S(IV) that is SO2∙H2O, HSO3-, and SO32- is given in the next figure. Note that the bisulfite ion is the most important for much of the range of cloud water. Recall that atmospheric CO2 creates cloud water with a pH of 5.6. The form of the sulfur becomes quite important when we think about reactions of sulfur compounds in the aqueous phase.
To find the pH of the solution, we can assume once again assume electroneutrality. Thus,
[H+] = [OH-] + [HSO3-] + 2 [ SO32- ]
[H+] = Kw/[H+] + HSO2 Ks1 pSO2/[H+] + 2 HSO2 Ks1 Ks2 pSO2 / [H+]2
or
[H+]3 – (Kw + HSO2 Ks1 pSO2) [H+] - 2 HSO2 Ks1 Ks2 pO2 = 0
By looking at the values for each of the terms, we can simplify this expression.
5.1.4. Ammonia /water equilibrium.
Ammonia is the major basic atmospheric constituent that enters aqueous solutions. It equilibrium equations are:
NH3 + H2O Û NH3 ·H2O
NH3 ·H2O
Û
NH4+ +
where NH4+ is the ammonium ion. The equilibrium constants has the following equations and values:

With these two equilibrium equations, we can find the ammonium ion concentration:
![]()
Thus, the total dissolved ammonia [NH3T] is given by:
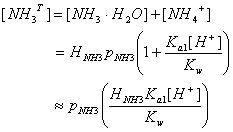
![]()
We were able to make the last approximation because for Ka1[H+]/Kw ~ 1.5x10-5 10-7 / 10-14 >> 1. Thus, for pH less than 8, [NH3T] ~ [NH4+]. For pH < 5, essentially all ammonia will be dissolved in the cloud water.

5.1.5. Nitric acid / water equilibrium.
After sulfuric acid, nitric acid is the most important acid that comes from pollution. It is through nitric acid that the oxidation processes that create ozone are linked to the processes that cause acid rain and aerosol formation. The equilibrium equations are:
HNO3 + H2O Û HNO3×H2O
HNO3×H2O Û NO3- + H+
The equilibrium constants and their values are:
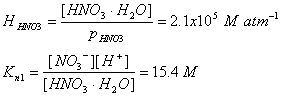
With these two equilibrium constants, we can find the nitrate concentration:
![]()
so that the total dissolved nitric acid is:
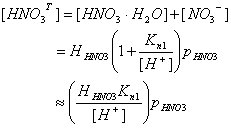
Kn1 = 15.4 M at 298 K. Thus, Kn1 / [H+] >> 1. Thus [NO3-] >> [HNO3×H2O]
The effective Henry’s Law constant is:
![]()
For a pH of 5, so that [H+] = 10-5 M, the effective Henry’s Law Constant is ~3x1013 M atm-1. Almost all of the nitric acid will be in the aqueous phase.
5.1.6. Hydrogen Peroxide / water equilibrium.
The equilibrium equations for hydrogen peroxide are:
H2O2(g) +H2O Û H2O2×H2O
H2O2×H2O Û HO2- + H+
The partitioning between H2O2×H2O and HO2- is given by:
![]()
![]()
![]()
We see that almost all dissolved H2O2 is in the form of H2O2×H2O for pH values that are typical for the atmosphere (i.e., less than 5.6).
5.1.7. Ozone.
Ozone is not very soluble in water. It exists essentially entirely in the aqueous form of O3∙H2O.
5.1.8. List of constants.
The following table contains the Henry’s Law Constants and the Equilibrium Constants for the atmospheric constituents and equilibria discussed above. These equilibria are temperature dependent, but we will assume that the temperature is always near 298 K. for our discussions.
|
Selected Henry’s Law Constants and Equilibrium Constants at 298 K. |
||
|
Equilibrium |
H (mole L-1 atm-1) |
K (M) |
|
H2O ↔ H+ + OH-
|
|
1.0 x 10-14 |
|
CO2 + H2O ↔ CO2∙H2O |
3.4 x 10-2 |
|
|
CO2∙H2O ↔ H+ + HCO3- |
|
4.3 x 10-7 |
|
HCO3- ↔ H+ + CO32- |
|
4.7 x 10-11 |
|
SO2 + H2O ↔ SO2∙H2O |
1.23 |
|
|
SO2∙H2O ↔ H+ + HSO3- |
|
1.32 x 10-2 |
|
HSO3- ↔ H+ + SO32- |
|
6.42 x 10-8 |
|
NH3 + H2O ↔ NH3∙H2O |
62 |
|
|
NH3∙H2O ↔ NH4+ + OH- |
|
1.8 x 10-5 |
|
HNO3 + H2O ↔ HNO3∙H2O |
2.1 x 105 |
|
|
HNO3∙H2O
↔ H+ + NO3- |
|
15.4 |
|
H2O2 + H2O ↔ H2O2∙H2O |
7.45 x 104 |
|
|
H2O2∙H2O ↔ H+ + HO2- |
|
2.5 x 10-12 |
|
O3 + H2O ↔
O3∙H2O |
0.011 |
|
5.2. Aqueous sulfur chemistry
The equilibrium processes descried above are reversible. If the partial pressure of SO2 decreases for some reason, like dilution or surface deposition, the sulfur in the aqueous phase will remain in equilibrium with the partial pressure and will thus decrease. The situation is quite different for more oxidized forms of sulfur, sulfates, the S(VI) compounds. These have a very low vapor pressure and remain for the most part in the liquid phase. Thus, the reactions that take S(IV) to S(VI) are essential for the production of acid rain.
S(IV) can be oxidized to S(VI) by a number of chemical species, including O3, H2O2, dissolved oxygen catalyzed by metals, and dissolved NO2.
Application to the atmosphere, which has clouds and fogs.
How do we apply liquid-phase chemistry to the atmosphere? How do we compare the rates of processes that occur in the liquid phase to those that occur in the gas-phase? Recall that the reaction rate is proportional to the concentration of the reactants. In the gas phase, we usually use the units molecules cm-3, while in the aqueous phase, we use the units M = moles/liter. Converting between these two can be a headache. However, for reactions or equilibria, it is the concentrations that matter. We can compare the gas-phase and aqueous phase rates by considering the rate in a volume of air. To calculate the rate for the aqueous phase, we need to know the volume of liquid that is in that volume of air. This value is usually called the liquid water content (LWC). In cloud physics, this is usually given units of g m-3. There is another value that is also used: the cloud water mixing ratio, which is wL = (volume of liquid)/(volume of air). The two are related. Since for water 1 m3 = 106 g, the conversion between the two is:
LWC = 106 wL
What are typical values for wL for different conditions? They are the following:
wet aerosol: 10-12 - 10-9
fog : 10-9 - 10-7
cloud : 5x10-8 - 3x10-6 , or 0.05 – 3 g m3.
Assume that a solute A is neither created nor destroyed. The distribution factor, fA, is the ratio of the aqueous phase concentration and the gas-phase concentration, in the same units (g/(Lof air)), or other units will do.
fA = (mass in liquid / volume of air)/(mass in air / volume of air) = concentration in the liquid in the volume of air / concentration in the gas phase.
We will denote the concentration in the liquid in the volume of air with CA(aq) and the concentration in the gas-phase with CA(g). Note that [A(aq)] is the concentration of A in the liquid in terms of moles/L(liquid).
So, CA(aq) = [A(aq)] wL, which has the units (moles/L(aq)) x (L(aq)/L(air)
CA(g) is different from [A(g)] because it must have units of moles/L(air) if fA is to be unitless.
Now, we can transform these relations using the Ideal Gas Law, to get the
gas-phase amount in terms of the partial pressure,
pA = CA(g) RT implies the CA(g) = pA / RT.
We should make sure that we use R = 0.08205 atm L mol-1 K-1 and pA in units of atm.
Further, we can transform the liquid phase concentration by using Henry’s Law:
CA(aq) = [A(aq)] wL = HA pA wL
We will need to use the appropriate Henry’s Law Constant, which in many cases will be the effective Henry’s Law Constant.
Putting all of this together gives us the ratio of A that is in the liquid phase to A in the gas phase:
![]()
What we are really interested in is the fraction of A that is in the liquid phase, denoted as XAaq:
![]()
For LWC = 10-9 l m-3, (R T LWC)-1 = 4x104 M atm-1. Thus, chemical species that have a Henry's Law constant, or effective Henry's Law constant much less than 4x104 M atm-1 will be mostly in the gas-phase, as in the figure. As a result, in the absence of oxidation, most SO2 will be in the gas-phase and most HNO3 will be in the liquid.

So, now we can put the aqueous phase reactions in the context of the atmosphere.
For a dissolved species A,
S(IV) + A(aq) ® products
In the liquid phase, the rate equation for this reaction is simply:
![]()
The units for this reaction rate are M s-1 = moles L-1 s-1. To get this reaction rate in terms of liters of air, we must multiply by wL. This will give us the reaction rate for A and S(IV) due to liquid phase the liquid phase reaction.
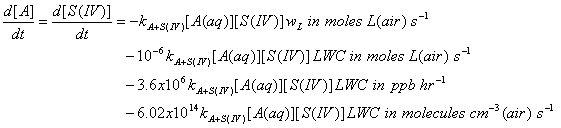
The reaction rate coefficient should have units of L mole-1 s-1; the concentrations are M = moles L(aq)-1; LWC is g m-3; and wL is (moles L(aq)-1)/(moles L(g)-1).
If we want to express these equations in terms of per cent change per hour, we simply divide the third expression by the mixing ratio of SO2 and multiply by 100:
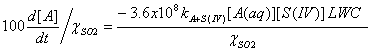
The lifetime is simply this 100 divided by this last equation, in units of hours.
We can also get the fraction converted per unit time. The fraction of sulfur converted per unit time is the conversion rate divided by the total amount of sulfur not yet converted, in both the gas and the liquid phase. The fractional rate is thus:
fraction converted per unit time = {d([S(IV)])/dt} / {pSO2/RT + H*SO2 pSO2 wL}
Now let’s look specifically at the oxidation of S(IV) to S(VI). We can see the reaction rates for typical amounts of different oxidants. The liquid-phase oxidants, H2O2 and O3, are largely created in the gas-phase and absorbed into the liquid. Note that O3 and H2O2 are both formed by processes involving HOx radicals. We should expect that the greatest oxidation will occur during the day, when oxidant concentrations are largest, and will be greater in the summer than the winter for the same reason.

Because the oxidation by HOOH is by far the largest for anything but very acidic or basic solutions, we will focus on it.
The reaction pathway is given by the equations:
HSO3-+H2O2 <==> SO2OOH- + H2O
SO2OOH- + H+<==> H2SO4
The specific rate equation is from Hofmann and Calvert (1985):
d([S(IV)])/dt = - {kHOOH+S(IV)[H+][H2O2(aq)][HSO3-] }/{1 + KHOOH[H+]}
where kHOOH+S(IV) = 7.5x107 M s-1, and KHOOH = 13 M-1. Note that we must determine [H+], [H2O2(aq)] = HH2O2 pH2O2. Note that we cannot use [S(IV)](aq), but must instead use [HSO3-], which can be calculated from the equilibrium conditions.
This reaction rate is essentially independent of pH when we multiply all the terms together. The reason for this weak dependence is that kHOOH+S(IV) is essentially independent of [H+], {H2O2(aq)] is independent of [H+], [HSO3-] depends on 1/[H+], and KHOOH[H+] << 1.
For pH2O2 = 1 ppb,
d[S(IV)]/dt = 300 mM h-1 (ppbSO2)-1 (700% SO2(g) h-1 (g water / m3 air)-1
The reactions on page 367 show that the immediate product is H2SO4, sulfuric acid.
What happens to it? It is ionized:
H2SO4(aq) Û HSO4- + H+ Ksvi1 = 1000;
HSO4- Û SO4 2- + H+ Ksvi2 = 1.02x10-2
H2SO4(g) Û H2SO4(aq) remains in liquid-phase.
Therefore, we can determine the dominant forms of S(VI):

Open and closed systems.
1. Open system.
1. Gas partial pressures are maintained at fixed values by resupply from new air or from photochemistry.
2. Not a very realistic condition.
2. Closed system.
- gas-phase partial pressures decrease with time.
- More representative of actual behavior.
- Masses of elements, such as S, N, C are conserved in the process.
Let us consider what happens in a closed system.
In an open system, [H2O2(aq)] = HH2O2 p0H2O2. In this case, the aqueous phase H2O2 is independent of LWC.
In a closed system, initial H2O2 is split between the gas and aqueous phases, as the cloud forms:
[H2O2]total = (pH2O2/RT) + [H2O2(aq)]closed wL
where recall that wL has units of L liquid / L
air and R = 0.082 (atm L)/(mole K).![]()
If we are in equilibrium, then Henry’s Law holds and:
[H2O2(aq)]closed = HH2O2 pH2O2
If initially there was no cloud, then the conservation equation for H2O2 becomes:
poH2O2 = pH2O2 + [H2O2(aq)]closed wL RT
= [H2O2(aq)] {(1/HH2O2) +wL RT}
Combining these equations, we get:
[H2O2(aq)]closed = (HH2O2 poH2O2) / (1 + HH2O2 wL RT)
Note that [H2O2(aq)] decreases as LWC or wL increases. More water means that more H2O2 can be accommodated in the aqueous phase. The open system is represented by the dashed lines. For the closed system, because pH2O2 is proportional to [H2O2(aq)], pH2O2 drops as [H2O2(aq)] drops. In addition, because pH2O2 is proportional to [H2O2(aq)], the fraction of H2O2 that is in the aqeuous form [H2O2(aq)] increases as the LWC increases.
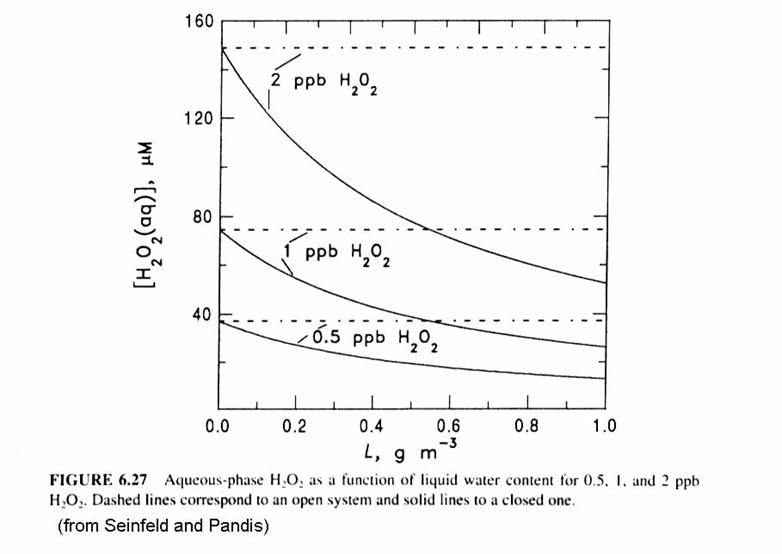
More sulfate is produced in the open system than the closed system, as in the figure below. This makes sense: the oxidants and SO2 are being constantly replaced and is thus able to continue the conversion of S(IV) to S(VI).

Low solubility gases, like ozone, maintain aqueous phase concentrations that are similar to that for open systems. For example, if wL = 10-6, T = 300 K, then HO3 wL RT = 0.0113 10-6 0.082 300 = 2.8 x 10-7, which is much smaller than 1 and can be ignored.
[O3(aq)]closed = (HO3 poO3) / (1 + HO3 wL RT) ~ (HO3 poO3)
We can do the same thing for dissociating species, such as HNO3:
[HNO3(aq)] +[NO3-] = (H*HNO3 pHNO3) / (1 + H*HNO3 wL RT)
where
H*HNO3 = HHNO3 {1 + Kn1 / [H+]}
Consider a cloud forming in clear air that contains SO2 and H2O2.
- Equilibrium is readily established.
- Concentrations of ions will adjust as the reactions proceed so that electroneutrality is maintained.
- The concentrations of all species can be defined by their equilibrium constants and partial pressures, except for sulfate and bisulfate, which are strictly in the aqueous phase.
- The reactions will proceed until either H2O2 or SO2 are depleted. If H2O2 is depleted first, then the SO2 will move further downwind and get oxidized to S(VI) by either gas-phase or aqueous phase processes downwind.
Global transport models suggest that gas-phase oxidation and liquid phase oxidation are about equally important for global SO2 emissions, even though the rates of oxidation in cloud water are several 10’s of percent per hour, while gas-phase oxidation is at most a few percent per hour. While clouds cover roughly half of Earth’s surface at any given time, they occupy a much smaller volume of the troposphere at any given time.
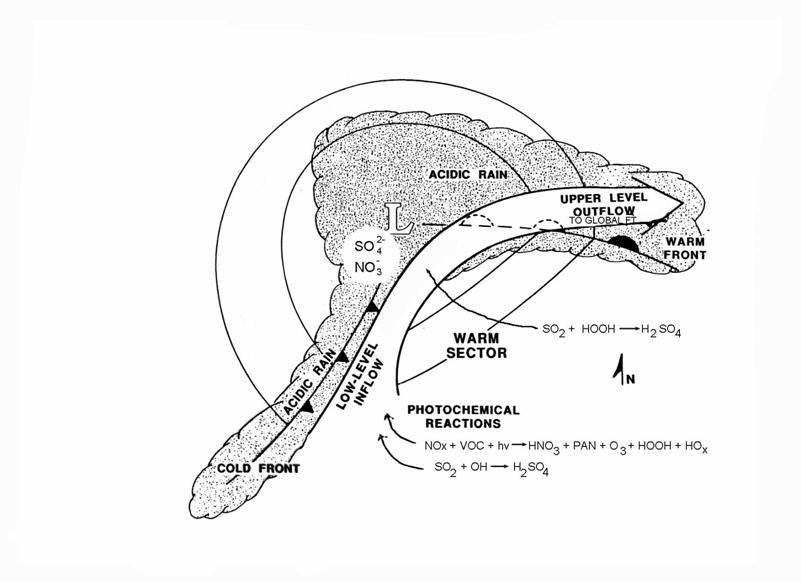
Once the sulfate aerosol is in cloud drops, it can come down as precipitation. The figure below depicts the processes that occur in a rain storm. The photochemical constituents are drawn up into the storm in the low level inflow. Aqueous phase conversion of SO2 to sulfate occurs. Rain out deposits some of this sulfate on Earth’s surface. What is not rained out can be delivered to the global free troposphere by the upper level outflow. This sulfate can then travel great distances as aerosol before it is brought down to lower levels by subsidence of it air parcel, where it can be deposited by dry or wet processes.
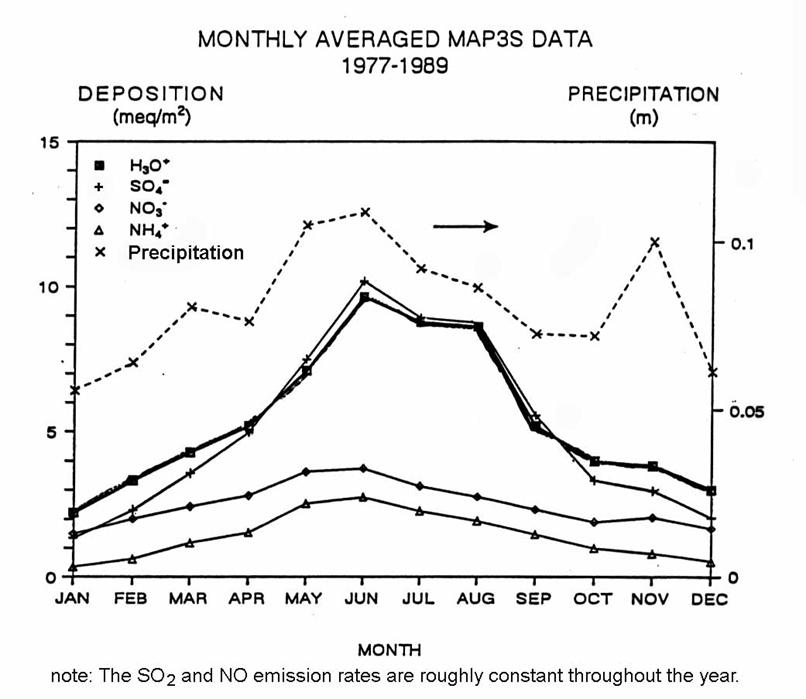
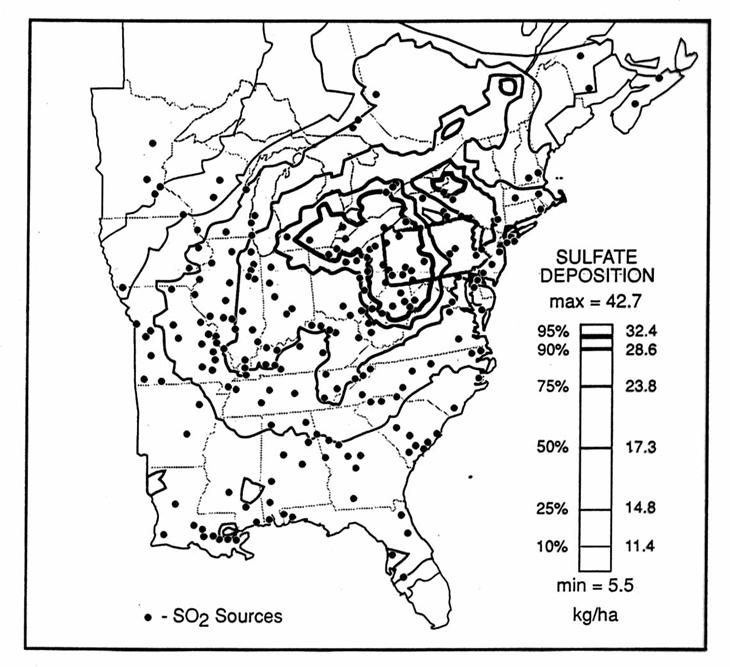
The pattern of sulfate deposition for the Eastern United States is shown in the following figure.

The figure below shows the annual variation of deposition for sites in the Eastern United States. There is a clear peak in deposition during the summer months. This peak is not due to increases in SO2 and NO emissions, which are roughly constant over the whole year. What could be the cause of this summertime increase?