The following article by Dr Marsha Singh, Queen's University Physics Department, is verbatim from:
http://physics.queensu.ca/~marsha/SAXSoverview.html
A Brief Overview of SAXS
Small angle x-ray scattering (SAXS) is a very well-established
measurement tool that has been
around for about 70 years. It is "special" in terms of the distinction between
SAXS and regular wide-angle x-ray
scattering by virtue of the location of the scattering of interest.
This is typically at small
angles in the vicinity of the primary beam and
extending to less than 2 degrees for standard wavelengths. The scattering
features at these angles correspond to structures ranging from tens to
thousands of angstroms.
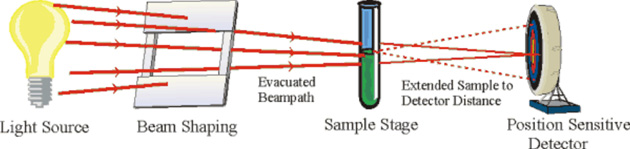
Figure 1: Schematic
SAXS Apparatus
Why SAXS?
Why do SAXS, instead of, say light scattering or electron
microscopy which might be considerably
more straightforward? In many cases, we need to look at bulk materials which are
opaque to visible light. The need to
produce very thin slices for electron microscopy can destroy the
very thing we want to look
at. Small angle neutron scattering is usually a possibility but certainly no more straightforward than
SAXS. In the cases described
in this document, SAXS is either the best or the only source of the information that we needed on
our materials.
How can SAXS be
performed?
The primary experimental ingredients to SAXS is the need for a well-collimated x-ray
beam with a small cross-section. Synchrotron
radiation sources with their intense brightness and natural
collimation are ideal when we consider the fact that most polymer
materials are very poor scatterers.
There is always some form of beam shaping
required to maintain the small
cross-section in going from the source to the sample and to reduce distortions from parasitic scattering from
whatever obstacles, including
air, are encountered. This is where much of the experimental effort is required. A sample stage that
may or may not involve heating elements, tensile stress apparatus, etc.,
then follows, ideally all within
an in-vacuum path. No preparation such as staining of the material is required, and thicknesses of between
1 and 3 mm are usually fine.
As shown here, and indeed in most
typical experiments, the SAXS technique is performed in transmission mode. In this mode, polymer samples
are typically 1-2mm thick, offering
about 63% absorbtion of the incident x-ray beam.
In situations where transimission mode operation is not a feasable
option, such as when the sample
of interest is a thin film on an opaque substrate of when only the surface microstructure is of interest,
one must resort to using a
combination of Grazing Incidence Diffraction geometry and SAXS, known as GISAXS. More information about GISAXS is
available at the link provided.
An extended sample-detector distance
is usually required to give
the barely scattered photons room to spread out from the main beam and also to reduce the detected x-ray background.
Finally, a position sensitive
detector, ideally 2-dimensional, is required to measure the scattered intensity. As sketched here, the
black spot would be the beamstop
that is absolutely essential to block
the main beam and the rings are a cartoon of the Debye rings one would see
from an ordered structure. As
we will see, most SAXS data is much less straightforward and appears as
a continuous function of scattering angle. For analysis, we will usually have to reduce this 2-D profile to a 1-D set
of intensity vs. angle data.
SAXS Data Analysis
Interpreting SAXS data can be a very difficult task unless
one is very lucky and the sample
fits one of the many idealized models that have been developed over the years. Regular WAXS tends to focus
on the location, width, shifts,
etc. of Bragg peaks which arise from crystalline lattice structures. One can still observe Bragg peaks
in SAXS but these will result from
regular spacings that are on the order of hundreds of Angstroms.
Most of the time, however, the observed curves
tend to be apparently featureless.
At very small angles, the shape of
the scattering in the so-called
Guinier region can be used to give
us an idea of the radius of gyration of any distinct structures that are on this type of lengthscale.
At higher angles, if we had a system
of relatively identical particles,
dilute enough for there to be no interactions, we might be able to see broad peaks that would also give
us information on the shape
of the particles. The sketch here showing Bragg peaks corresponds to a system of strongly interacting particles
which would obscure this type of single-particle information.
At still higher angles, the so-called
Porod region, the shape of the curve is useful in obtaining information
on the surface-to-volume ratio of the scattering
objects. This can also be used to can information on the dimensions
of our scattering particles.
Finally, the area under the curve gives
us the so-called INVARIANT
which is a measure of how much scattering material is seen by our beam. Changes in the invariant are useful
in monitoring the crystallization
process in polymer materials.
All of these are so-called DIRECT methods
of analysis which give us information
based on interpretation of the clean (background corrected) data with no further manipulation.
However, all of these parameters
are based on well-defined assumptions such as the existence of uniform density within our so-called particle,
uniform density in the background,
sharp interfaces between the two, etc. We can go further when these approximations do not apply by
fourier transforming the data
to get real space information (such as obtainable by electron microscopy). We've used the GNOM routine
provided by a Russian colleague
for this purpose.
We can also propose specific structures,
calculate the scattering, then
fit the data to obtain the parameters
defining our model structure. This tends to imply that we already know the
answer, not the general case.
Finally, when we don't really have
any clear order to base our
interpretation on, we can assume a strongly DISORDERED structure and turn to fractal analysis (where
the disorder is itself a form of order)
or paracrystal analysis (where the system is really just a heavily distorted regular structure).
Further Reading
·
M.A. Singh and
C. Barberato, Small-Angle X-ray Scattering from Soft Materials, Physics in Canada, September/October 1997.
·
O. Glatter and O. Kratky, Small
Angle X-ray Scattering, New York: Academic Press, 1982.
·
L.A. Feigin and D.I. Svergun, Structure
Analysis by Small-Angle X-ray
and Neutron Scattering, New York: Plenum Press, 1987.